Juvenile xanthogranuloma
JXG; juvenile xanthogranuloma; non-X histiocytosis of childhood
Juvenile xanthogranuloma is the commonest form of paediatric non-Langerhans cell histiocytosis — a benign self-limiting proliferation of histiocytes typically presenting as a yellow-orange to red-brown papule or nodule on the head, neck or upper trunk of an infant or young child. Most JXGs (90%) appear before age 2 and spontaneously resolve over 1–6 years, often leaving residual telangiectasia or atrophic scarring. Extracutaneous involvement is uncommon (< 5%) but clinically critical — particularly ocular JXG (iris involvement → uveitis, hyphaema, glaucoma) and the recognised association with neurofibromatosis type 1 and juvenile myelomonocytic leukaemia (the JXG-NF1-JMML triad).
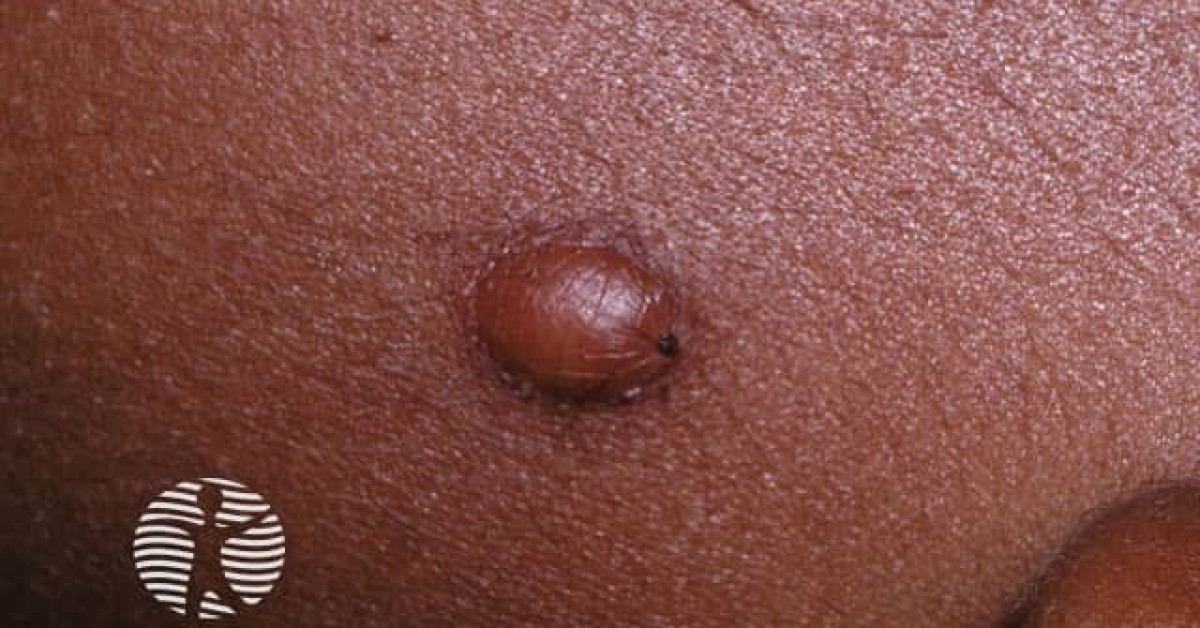
Clinical features
- Yellow-orange to red-brown firm papule or nodule, 2–20 mm; occasionally larger.
- Sites — head, neck, upper trunk (commonest); any site possible.
- Solitary in 60–80%; multiple in 20–40%.
- Onset — 70% within first year, 90% by age 2; rare adult-onset cases (different biology).
- Self-resolution over 1–6 years with residual hypopigmentation, atrophy or telangiectasia.
- Asymptomatic.
Extracutaneous involvement
- Ocular JXG — most clinically important extracutaneous site; iris involvement leads to uveitis, hyphaema, glaucoma. Examine eyes carefully in any infant with multiple JXG; specialist ophthalmology referral.
- Pulmonary, hepatic, splenic, CNS involvement — rare but described.
- Bone marrow involvement — rare; consider JMML.
- Most extracutaneous disease resolves with the cutaneous lesions, but ocular disease may require treatment.
JXG-NF1-JMML triad
- Children with JXG (especially multiple) have an increased risk of neurofibromatosis type 1.
- Children with both JXG and NF1 have a 20–32× increased risk of juvenile myelomonocytic leukaemia (JMML) — though the absolute risk remains low.
- Workup — full skin examination for café-au-lait macules + axillary / inguinal freckling (NF1 features); CBC with manual film at baseline and as clinically indicated; haematology referral if cytopenia or atypical cells.
- This triad is the principal reason to take JXG seriously beyond the typical benign cutaneous course.
Histology
- Dermal infiltrate of foamy / vacuolated histiocytes.
- Touton giant cells — multinucleated cells with peripheral nuclei in a wreath and foamy cytoplasm — highly characteristic.
- Mixed inflammatory infiltrate — lymphocytes, eosinophils.
- Immunohistochemistry — CD68+, factor XIIIa+, fascin+; S100 / CD1a / Langerin negative (distinguishes from Langerhans cell histiocytosis).
- BRAF V600E mutation absent (contrasting with LCH).
Management
- Cutaneous JXG — observation; reassurance about spontaneous resolution.
- Excision occasionally for diagnostic uncertainty, cosmetic concern or rapid growth.
- Examine all children with JXG for NF1 features (café-au-lait, freckling) and consider ophthalmology review.
- FBC for new multifocal JXG in NF1 child; refer haematology if any haematological abnormality.
- Ophthalmology screening every 6 months in NF1 children with multiple JXG until age 5.
- Ocular JXG — topical / systemic steroid; methotrexate; cyclosporin; vitrectomy.
- Disseminated systemic JXG — chemotherapy regimens analogous to LCH.
References
- Janssen D, Harms D. Juvenile xanthogranuloma in childhood and adolescence — a clinicopathologic study of 129 patients from the Kiel pediatric tumor registry. Am J Surg Pathol; 2005.
- Cypel TK, Zuker RM. Juvenile xanthogranuloma — case report and review. Can J Plast Surg; 2008.
- Burgdorf WH, Zelger B. JXG, NF1 and JMML — the triple association. Pediatr Dermatol; 2004.
Spot a correction?
If any clinical statement, citation or link on this page needs updating, please email admin@skinoncology.net with the page name, the proposed correction and the supporting source.